
Understanding H2 binding and activation is vital within the context of designing transition metallic catalysts for a lot of processes, together with hydrogenation and the interconversion of H2 with protons and electrons.
This work experiences the primary thermodynamic and kinetic H2 binding studies for an isostructural collection of first-row metallic complexes: NiML, the place M = Al (1), Ga (2), and In (3), and L = [N(o-(NCH2PiPr2)C6H4)3]3-.
Thermodynamic free energies (ΔG°) and free energies of activation (ΔG‡) for binding equilibria have been obtained by way of variable-temperature 31P NMR studies and lineshape evaluation.
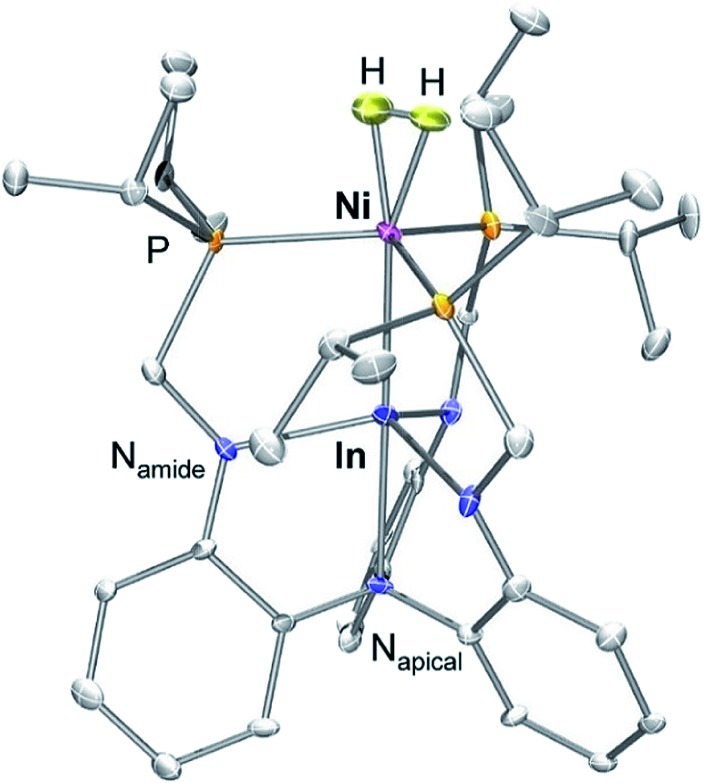
The supporting metallic exerts a massive affect on the thermodynamic favorability of each H2 and N2 binding to Ni, with ΔG° values for H2 binding discovered to span almost the complete vary of earlier experiences. The non-classical H2 adduct, (η2-H2)NiInL (3-H2), was structurally characterised by single–crystal neutron diffraction-the first such examine for a Ni(η2-H2) complicated or any d10 M(η2-H2) complicated.
UV-Vis studies and TD-DFT calculations recognized particular digital structure perturbations of the supporting metallic which poise NiML complexes for small–molecule binding. ETS-NOCV calculations point out that H2 binding primarily happens by way of H-H σ-donation to the Ni 4p z -based LUMO, which is proposed to develop into energetically accessible because the Ni(0)→M(iii) dative interplay will increase for the bigger M(iii) ions.
Linear free-energy relationships are mentioned, with the activation barrier for H2 binding (ΔG‡) discovered to lower proportionally for extra thermodynamically favorable equilibria. The ΔG° values for H2 and N2 binding to NiML complexes have been additionally discovered to be extra exergonic for the bigger M(iii) ions.
Building Blocks for High-Efficiency Organic Photovoltaics. Interplay of Molecular, Crystal, and Electronic Properties of Post-Fullerene ITIC Ensembles.
Accurate single–crystal x-ray diffraction knowledge provide a distinctive alternative to evaluate and distinction the atomistic particulars of bulk heterojunction photovoltaic small–molecule acceptor structure and packing, in addition to present a necessary start line for computational digital structure and cost transport evaluation.
Here we report diffraction-derived crystal constructions and computational analyses on ITIC and seven derivatives (together with three new crystal constructions: ITIC-C3, m-ITIC-C6, and ITIC-C4-4F. IDTT acceptors sometimes pack in a face-to-face style with π-π distances starting from 3.28-3.95 Å. Additionally, edge-to-face packing is noticed with S⋯π interactions as quick as 3.21-3.24 Å.
ITIC finish group identities and aspect chain substituents affect the character and power of noncovalent interactions (e.g. H-bonding, π-π) and correlate with the noticed packing motif, digital structure, and cost transport properties of the crystals. Density useful principle (DFT) calculations reveal comparatively massive nearest-neighbor intermolecular π-π digital couplings (5.85-56.eight meV) and correlate the character of the band structure with the dispersion interactions within the single crystals and core-end group polarization results.
This mixed experimental and theoretical work reveals key insights into crystal engineering methods for indacenodithienothiophene (IDTT) acceptors, in addition to normal design guidelines for natural photo voltaic cell post-fullerene small molecule acceptors.